

What is Thalassemia?
The thalassemias are a group of disorders that prevent the body from producing a sufficient quantity of high quality blood. This condition is caused by changes to the genes that control the production of haemoglobin. Haemoglobin is a protein in red blood cells that carries oxygen around the body-changes affecting haemoglobin result in severe anaemia.
The human body relies on blood with normal haemoglobin to function well.
Thalassemia is an inherited genetic disease that is passed from parent to child through the genes. Both male and female children have equal chances of getting the disease. It is not transmitted through blood, air or water, or through physical or sexual contact with a patient, nor can it be caused by poor nutrition or medical conditions.
Each person has two of every kind of gene, one passed on from the mother, the other from the father. Among other genes, these two genes that control how haemoglobin is formed is found in each red cell. Now consider three sorts of married couples:
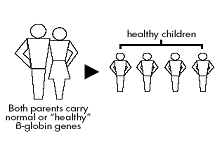
- Both parents are normal : They cannot possibly pass on thalassemia trait or thalassemia major to their children.
 |
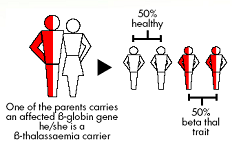
- One parent has a thalassemia and one is normal For each child there is a one in two (50%) chance of inheriting the thalassemia gene from the carrier parent: if this happens the child will have thalassemia trait. There is also a one in two (50%) chance of inheriting the normal gene from the carrier parent: if this happens the child will be completely normal. None of this couple’s children can have thalassemia major.
 |
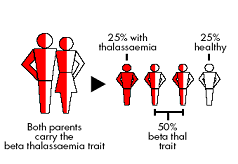
- Both parents are thalassemia carriers : i.e they are a “couple at risk” The couple who are both carriers have a one in four (25%) chance in each pregnancy of having a child with thalassemia major; a one in two (50%) chance of having a child with thalassemia trait; and one in four (25%) chance that the child will inherit a normal gene from both parents and so will be completely normal.
 |
Types of Thalassemia
- Thalassemia Major-thalassemia patients.
- Thalassemia Minor-carrier, asymptomatic and healthy.
How does a patient with Thalassemia Major patient present?
The red blood cells of a person with thalassaemia major can only survive for a few weeks, compared to normal red blood cells that survive for around four months. Lack of haemoglobin results in reduced oxygen to every cell in the body.
The following signs appear in early childhood:
- Child appears normal at birth. However, he/she will slowly become pale (anaemic), more severe and obvious when she/he reaches the age of 6-18 months.
- Child is often weak and restless.
- Severe anaemia can cause breathing difficulties.
- Jaundice.
- Poor appetite.
- Distended abdomen due to swelling of liver and spleen.
- Changes in facial bone structures.
How to diagnose Thalassemia?
- History and physical examination.
- Laboratory investigations:
- Haematological indices. These haematological parameters are measured by electronic equipment – a red cell counter. It assesses the size and volume of red blood cells and the amount of haemoglobin contained in them. Thalassaemia is diagnosed when the size and volume of red blood cells and the concentration of haemoglobin inside them are significantly reduced.
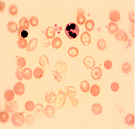
- Blood film and RBC morphology. Observed under a microscope, the red blood cells appear paler (hypochromic) and smaller (microcytic) than normal and – very importantly – the majority have abnormal shapes: anisocytosis, poikilocytosis.
 |
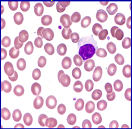 |
| Blood film of Thalassemia patient | Normal blood film |
- Haemoglobin electrophoresis. This is a process that separates the different proteins that make up a haemoglobin molecule – i.e. HbA,HbA2, and HbF. A diagnosis of thalassaemia is indicated where levels of foetal haemoglobin(HbF) are higher than normal and may vary between 20-90%. HbA2, which usually accounts for up to 3% of normal adult haemoglobin, may be non-existent, reduced, normal or slightly elevated.
- Molecular methods. These are specialised ways of confirming or obtaining more specific information in a diagnosis, using DNA investigation.
How do we treat Thalassemia?
Thalassemia major patients needs to undergo 2-4 weekly blood transfusion. This will correct the anaemia, and enable the body tissues to get normal amount of oxygen. This allows the patient to live and grow normally. It lets bone marrow rest so the bones can develop normally and will be strong and the face will look normal.
- Regular blood transfusions will cause the accumulation of iron in the vital organs such as heart, liver and endocrine glands. This eventually results in multiple organ dysfunction.
- Excessive iron can be removed by chelation therapy. Iron chelators can be given through injection or orally.
- Splenectomy (removal of spleen) may be necessary in certain situation.
- Bone marrow transplant may be necessary provided there is a suitable donor among family members.
- Gene therapy is still in experimental phase.
Iron chelators
Regular blood transfusions can lead to a build-up of iron and this can cause serious side effects including diabetes, heart failure and liver disease.
Medications can be used to remove excess iron from the blood. These medications are called ‘iron chelators’ – they work by binding with the iron and allowing it to be excreted by the kidneys.
The most commonly used medication is injected using a slow infusion pump that runs over several hours.
Oral iron chelators (tablets) may be prescribed when the infusion treatment is unsuitable or ineffective. However, these medications may have severe side effects and must be carefully monitored.
Injection-desferal
 |
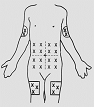 |
Oral- L1and ICL 670
 |
 |
What are the complications if Thalassemia is not treated?
If not treated, thalassemic patients will develop all the described symptoms and might die due to severe anaemia.
Although multiple lifelong transfusions save lives, they also cause serious side effects due to iron overload in the bodies of thalassemic patients. Over time, people with thalassemia accumulate deposits of iron in their bodies, especially in the liver, heart, and endocrine (hormone-producing) glands. The deposits eventually can affect the normal functioning of the heart, and liver, in addition to delaying growth and sexual maturation. These complications can be prevented with regular iron chelation.
Survival and quality of life in Thalassemia patients
Dramatic improvements in the clinical management of thalassaemia major over the last 10-20 years have led to an impressive increase in the lifespan and quality of life of patients who have access or can afford and comply to recommended treatment regimes.
Can Thalassaemia be prevented?
Thalassaemia can be prevented by carrier screening and avoiding marriages among Thalassemic carriers.
Are there any support groups for the care of Thalassaemia children?
Contact the nearest hospital, clinic or Thalassemia Association.
| Last reviewed | : | 26 April 2012 |
| Content Writer | : | Dr. Zulaiha Muda |
| Reviewer | : | Dr. Hishamshah b. Mohd Ibrahim |





