

Introduction
Inborn Errors of Metabolism (IEM) comprises a group of genetic defects that affects the metabolic pathways in our body. It is due to defects in genes that produce enzymes or cofactors, which are essential in many biochemical reactions for proper functions of organs and energy production for maintaining health of our body. The lack of a particular enzyme can cause a block in a biochemical reaction, which may lead to accumulation of toxic substances (substrate) and reduced ability in production of essential compounds (products), thereby interfering in the normal function of the organ, such as the brain, nerve, liver, heart, eye, muscle, bone, or other organs. Both situations will result in adverse health consequences1,2.
There are more than 400 types of IEM diseases identified. Some of the common IEMs detected in Malaysia include glucose-6-phosphate dehydrogenase deficiency (G6PD), maple syrup urine disease (MSUD), methylmalonic aciduria (MMA), propionic aciduria (PPA), urea cycle defects (UCD), phenylketonuria (PKU), mucopolysaccharidosis (MPS) and mitochondrial disease.
The overall incidence of IEM is 1 in 4000 live birth3 and is affecting all races in the world. IEM usually affects babies and children but they can also occur in adulthood.
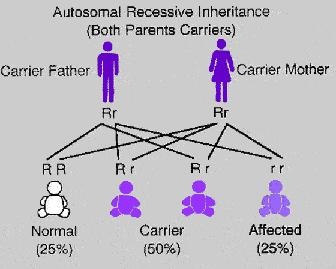
All IEMs are transmitted either by autosomal recessive or X-linked. The affected child inherited two defective genes each from one parent. The parents are usually healthy because they are carriers of the defective gene. The inheritance is called autosomal recessive. The risk of the particular couple to have another child in future with the same disease is 25%.
Sign And Symptoms
Some of the common sign and symptoms are poor feeding, frequent vomiting, seizure, lethargy from a short well period after born, hypotonia, failure to thrive and loss of conciousness. However, the clinical presentation depends on severity of the enzyme deficiency and clinical group of IEM.
The first group of IEM which causes acute or progressive intoxication from the accumulation of toxic compounds such as amino acid disorders (e.g., MSUD and PKU), organic acid disorders (MMA or PPA) and sugar intolerance (galactossemia, hereditary fructose intolerance). Common presenting features are poor feeding, vomiting, lethargy, septicaemic-like illness, peculiar smell, seizure, metabolic acidosis and hyperammonemia which will proceed to coma and death if not diagnosed and treated. Mild disorders usually present with mild symptoms such as developmental delay and failure to thrive.
The second group comprises disorders due to defects in energy production such asglycogen storage diseases, gluconeogenesis disorders and fatty acid oxidation disorders. The affected individual commonly presents with growth failure, hypoglycemia, hypotonia and reduced fasting tolerance.
The third group includes disorders with abnormal function of various organelles such as lysosome, peroxisome and mitochondrion. Patients usually present with progressive multisystemic disease with organ enlargement, facial dysmorphism and mental retardation2.
Complication
Early diagnosis and appropriate treatment given, the clinical and neurological effects may be prevented in 75% of these children4. If delayed in diagnosis and treatment, many of the affected children will require intensive care, be hospitalised for extensive periods, and will need long-term institutional care.
Management
Depending on the type of IEM, various forms of treatment can offered:
- Dietary restriction
- Vitamin supplement
- Enzyme replacement therapy
- Bone marrow transplant
- Liver transplant
Diagnosis Techniques
IEM are usually diagnosed by blood or urine test.
Screening Test:
- Newborn screening for amino acids disorders, organic acidurias and fatty acids oxidation defects.
- Total Galactose and GALT for Galactosemia.
- Urine cystine for cystinuria.
- Multiplex enzyme assay for screening of lysosomal diseases
Definitive tests
- Amino acids analysis
- Urine organic acid profiling
- Plasma Carnitine and acylcarnitines analysis
- Purine and pyrimidine analysis
- Glycosaminoglycans and Oligosaccharides detection
- Enzyme activities assay
- Mutation analysis
Prevention
- To screen baby at 48 to 72 hours of life before the baby become sick and deteriorate
- Only 3 drops of blood need for newborn screening and able to detect up to 30-40 types of IEM diseases
- Early diagnosis and prompt treatment in high-risk baby and prevent mental retardation and physical inability
- Genetic counseling can be provided
Support group
- Malaysian Metabolic Society www.mms.org.my
- Malaysian Lysosomal Diseases Association. www. mymlda.com
References
- Inborn Metabolic Diseases. 5th ed. Springer, 2012.
- The Metabolic and Molecular Bases of Inherited Disease. 7th ed. McGraw-Hill, 2000.
- Debra L W. Inborn Errors of Metabolism. http://emedicine. Medscape.com/article/804757-overview#a0199 . Updated: March 13 2012
- Novello AC. Inherited Metabolic Diseases: collaborating for the health of all children. Biochem Med Metab Biol 1993; 49:277-284.
 |
 |
 |
| Last Reviewed | : | 3 January 2014 |
| Writer | : | Chen Bee Chin |





